Library QC
Proximity ligation properties
At step Removing PCR duplicates you used the flag --output-stats, generating a stats file in addition to the pairsam output (e.g. –output-stats stats.txt). The stats file is an extensive output of pairs statistics as calculated by pairtools; including total reads, total mapped, total dups, and total pairs for each pair of chromosomes. Although you can directly use the pairtools stats file as is to get informed on the quality of the Omni-C® or Micro-C® library, we find it easier to focus on a few key metrics. We include in this repository the script get_qc.py that summarize the paired-tools stats file and presents them in percentage values in addition to absolute values.
The figures below outline how the values on the QC report are calculated:
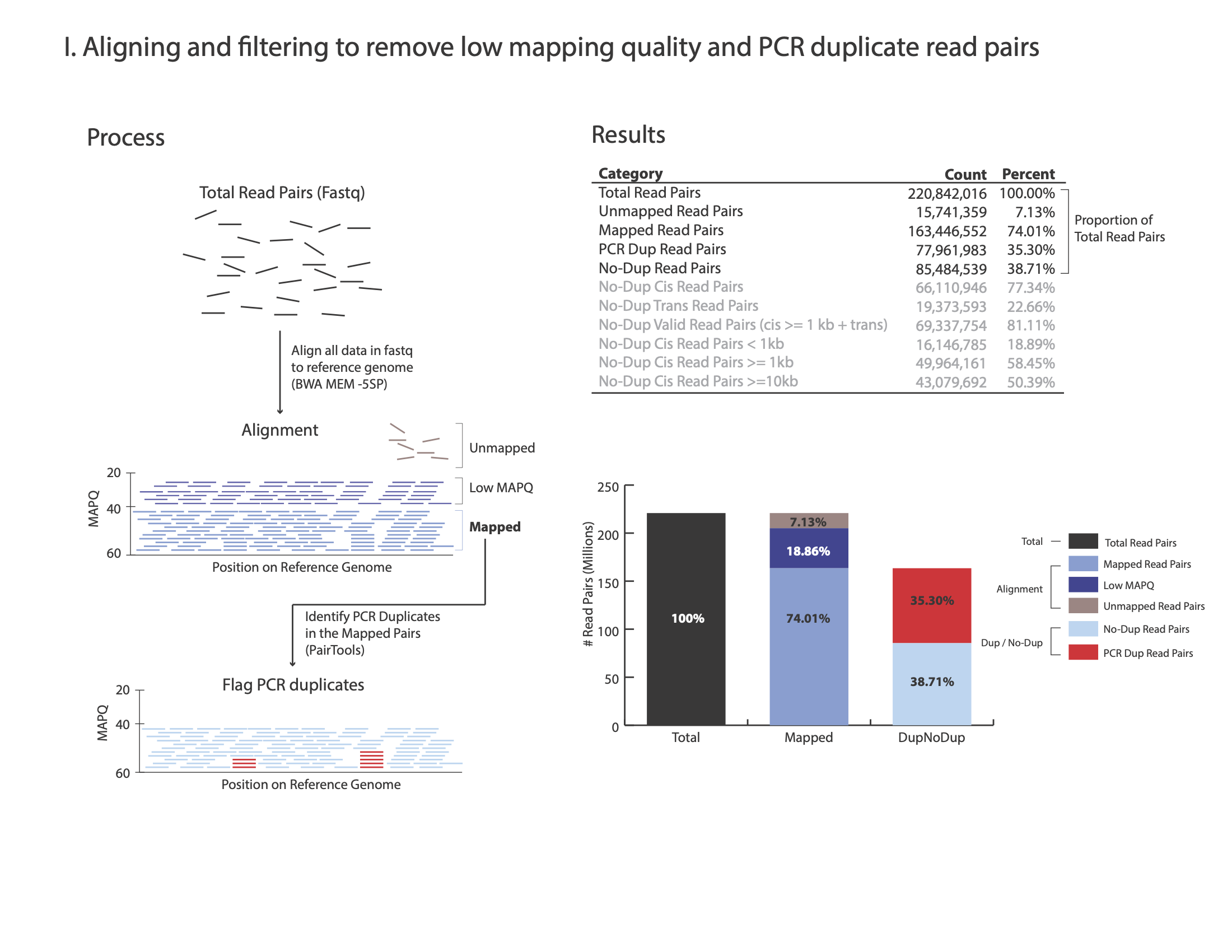
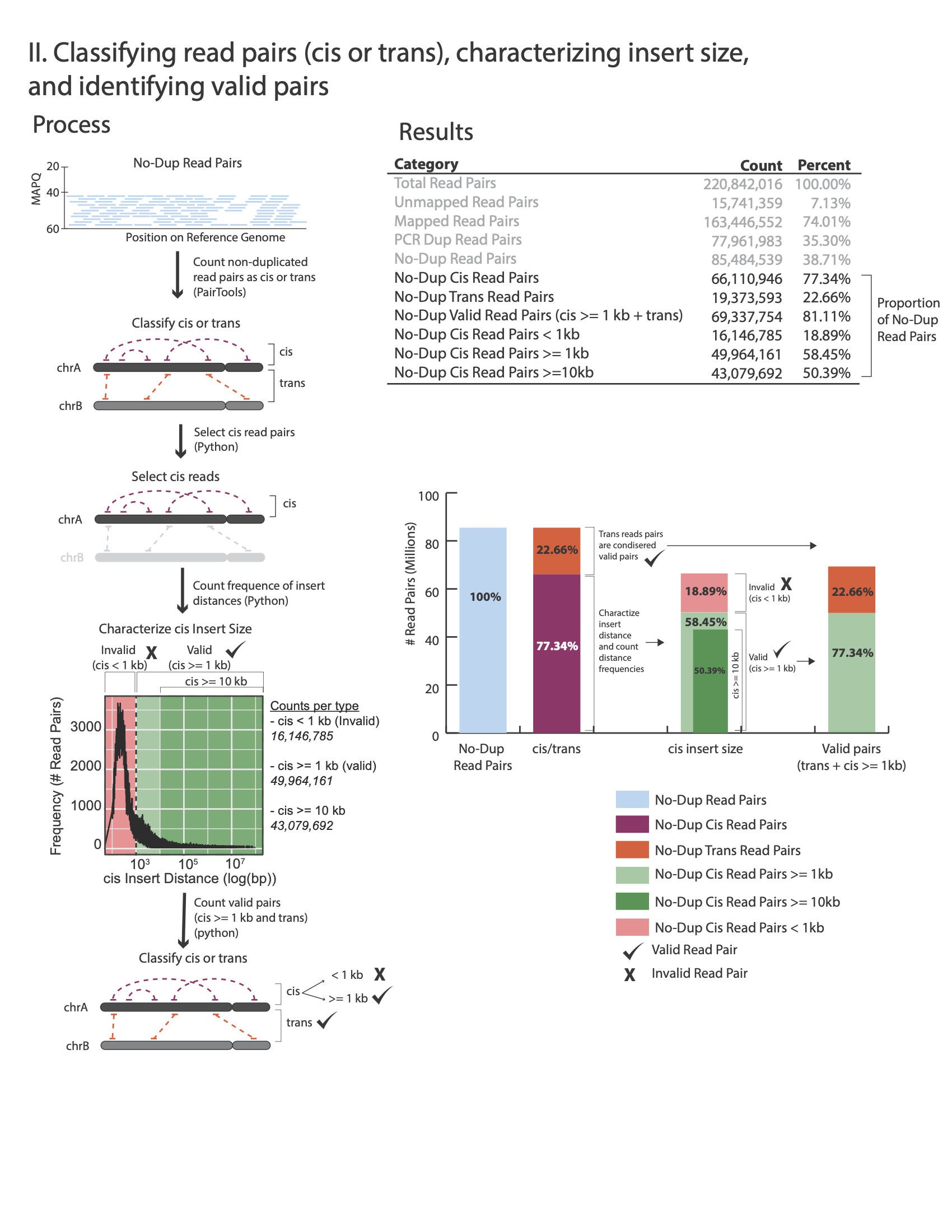
Command:
python3 ./capture/get_qc.py -p <stats.txt>
Example:
python3 ./capture/get_qc.py -p NSC_rep1_stats.txt
After the script completes, it will output (if you ran the NSC rep1 sample you should expect to generate these same results):
Total Read Pairs 253,341,035 100%
Unmapped Read Pairs 14,393,599 5.68%
Mapped Read Pairs 195,566,613 77.2%
PCR Dup Read Pairs 51,471,702 20.32%
No-Dup Read Pairs 144,094,911 56.88%
No-Dup Cis Read Pairs 94,499,595 65.58%
No-Dup Trans Read Pairs 49,595,316 34.42%
No-Dup Valid Read Pairs (cis >= 1kb + trans) 125,160,568 86.86%
No-Dup Cis Read Pairs < 1kb 18,934,343 13.14%
No-Dup Cis Read Pairs >= 1kb 75,565,252 52.44%
No-Dup Cis Read Pairs >= 10kb 46,482,376 32.26%
For each library, we recommend a minimum of 150 M Total Read Pairs
Typically, PCR Dup Read Pairs range from 10% up to 35%
No-Dup Trans Read Pairs are typically around 20% to 30% but lower or higher values alone are not reflective of poor library quality (e.g. 12%-35% are often observed with good quality libraries)
No-Dup Cis Read Pairs >= 1kb is typically between 50% to 65%
Target enrichment QC
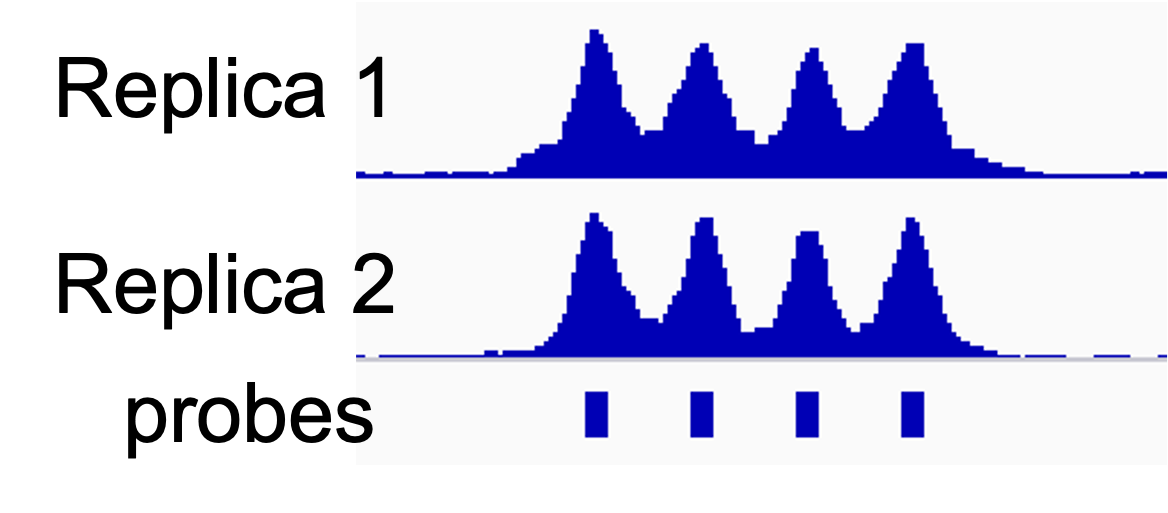
To evaluate the level of target enrichment we will compare the coverage over the probes (targeted regions) and the overall fraction of the captured reads in our library.
On-target rate
We define the on-target rate as the percentage of read pairs that map to targeted regions. The on-target rate uses read pairs and not reads since due to proximity ligation, half of the read-pair is expected to map elsewhere in the genome, such that only one read from the pair maps to the targeted region.
Since DNA fragments can extend beyond the sequenced region and as little as a 30 bp match is sufficient for capture, we treat reads that fall in close proximity to the targeted regions (up to 200 bp upstream or downstream from a probe) as on-target reads.
To count the number of on target pairs we will use:
*PT.bam file that you generated in the previous section (not the CHiCAGO compatible bam)
Bed file with the probe positions with 200 on both sides (the files h_probes_200bp_v1.0.bed for human and m_probes_200bp_v1.0.bed for mouse can be found in the Data sets section).
Count on-target read pairs:
Command:
samtools view <*PT.bam file> -L <padded bed file> -@ <threads> \
|awk -F "\t" '{print "@"$1}'|sort -u|wc -l
Example:
samtools view NSC_rep1.PT.bam -L h_probes_200bp_v1.0.bed -@ 16|awk -F "\t" '{print "@"$1}'|sort -u|wc -l
Samtools view with the -L argument enables the extraction of only the reads that mapped to the region of interest. The awk command helps us parse the file and extract the read ID information. The sort command with a -u (unique) argument will remove any multiple occurrences of the same read ID (to avoid counting read1 and read2 of the same pair if both mapped to the target region). And finally, wc -l counts the read IDs in this list.
The example above will output the value: 93,171,111 (On-Target Read Pairs)
There is no need to count the total read pairs in the bam file (which represents the total number of pairs, or 100%) as it was already reported by the QC script above, labeled as No-Dup Read Pairs (in our example: 144,094,911).
Now you can calculate the on-target rate:
And in the example above:
The on-target rate of the NSC replica1 example library is 64.7%. This is a typical on target rate, although occasionally lower values may be observed (as low as 40%).
Coverage depth
There are multiple methods and tools that enable the calculation of coverage depth from a bam file at different regions. We chose to use the tool mosdepth as we find it to be easy to use and relatively fast.
Use the probe bed file (and bait bed file if desired) to calculate coverage using the position sorted bam file (e.g. mapped.PT.bam. Do not use the CHiCAGO compatible bam file):
Command:
mosdepth -t <threads> -b <bed file> -x <output prefix> -n <bam file>
Example:
mosdepth -t 16 -b h_probes_v1.0.bed -x NSC_rep1_probes -n NSC_rep1.PT.bam
This command will yield multiple output files. Specifically, two files useful for QC-ing your libraries are: a) a bed file detailing the mean coverage per region (region being probe location, based on the input bed file), e.g. NSC_rep1_probes.regions.bed.gz and b) a summary output file, e.g. NSC_rep1_probes.mosdepth.summary.txt. The summary file provides information on the mean coverage of the total genome (second to last row) and mean coverage of the total_region (targeted region of interest - the last row in the summary). To print the header and two last summarizing rows, follow this example:
head -1 NSC_rep1_probes.mosdepth.summary.txt;tail -2 NSC_rep1_probes.mosdepth.summary.txt
This will output the following:
chrom length bases mean min max
total 3088269832 39020721947 12.64 0 482767
total_region 19337280 7835787504 405.22 0 8129
In this example (NSC rep1), the mean coverage over targeted regions is 405.22, while non-targeted regions have a mean coverage depth of only 12.64. Overall, the coverage depth is 32 times higher at targeted regions vs non-targeted regions: \(405.22/12.64 = 32\). The fold difference between the mean coverage depth of targeted regions and non-targeted regions is typically around 30, just as seen in this example.
The bed files with mean coverage values at on-target regions (e.g. NSC_rep1_probes.regions.bed.gz and NSC_rep2_probes.regions.bed.gz) will be used to assess replica reproducibility.
 Running the QC steps can be completed in less than 2 hours on an Ubuntu 18.04 machine with 16 CPUs, 1TB storage and 64GB memory.
Running the QC steps can be completed in less than 2 hours on an Ubuntu 18.04 machine with 16 CPUs, 1TB storage and 64GB memory.